ORFanage¶
ORFanage aids in finding the best matching OpenReadingFrame for each transcript in the GTF file based on evidence from one or more reference annotaitons. The method is designed to identify cases of known ORFs fitting the query transcript both with and without modifications, introduced by additional exons, alternative start and end sites, etc. ORFanage is also designed to quantify any changes to the reference annotation which are introduced by the splice variation.
Preparing input data¶
There are no special requirements for the input files. To run an analysis at the minimum you will need
GTF/GFF file with a set of transcripts you wish to annotate
GTF/GFF file with a set of annotated proteins to be used
Sometimes, transcripts in the files at your dispaosal may contain synonymous transcript type such as mRNA for “transcript”, etc. ORFanage will discard such records and fail to annotate them properly.
To correct any inconsistencies with the input files, we recommend you first run gffread software on your data [1]. Here is a sample command which creates
a standardized GTF file (-T), adjusts stop codon position (--adj-stop) and discards any transcripts with incomplete or invalid ORFs (-F, -J).
$ gffread -g genome.fa --adj-stop -T -F -J -o corrected.gtf input.gtf/.gff3
Here is a set of other utilities you may find helpful in generating and standardizing your input files [2]:
1$ bedToGenePred input.bed12 gene_pred.gpf
2$ genePredToGtf "file" gene_pred.gpf output.gtf
ORFanage produces results in GTF format. Other formats (such as BED12 or GFF3) may be supported in the future but are unavailable at the moment. To convert results back into the desired format you may find the following commands useful:
1$ gtfToGenePred input.gtf gene_pred.gpf
2$ genePredToBed gene)pred.gpf output.bed12
Annotating Transcripts with ORFanage¶
At the minimum ORFanage will annotate
$ orfanage --reference genome.fa --query query.gtf --output output.gtf template.gtf
Multiple template annotations can be passed to ORFanage in the same command either as space-delimited list of files
$ orfanage --reference genome.fa --query query.gtf --output output.gtf template.1.gtf template.2.gtf template.3.gtf
or by providing a file with a list of template annotations (GTF/GFF3) files. This option is particularly useful when a large number of reference annotations is to be used together.
$ orfanage --reference genome.fa --query query.gtf --output output.gtf template.lst
Other optional arguments may be provided in the command before the list of template annotations. These parameters can control sensitivity, speed, outputs and other features and are described extensively below.
Output Formats¶
ORFanage is primarily designed to anotate ORFs for transcripts included in a query GTF file. The main output file generated by the software is a a modified copy of an input annotation with ORFs included as additional records of a CDS type.
Note
GTF/GFF3
If a GFF-formatted file is provided as an input to ORFanage the format will be converted into GTF specifications in the output. Future versions of the software will likely support multiple additional formats such as GFF3, BED12, etc. However, at the moment only GTF is supported for the main output of the method.
ORFanage can also output extended information regarding all ocmparisons and candidate ORFs computed during the run. This information can be valuable in searching for specific differences between query and reference transcripts. It can also be used to find alternative translations as well as to examine ORFs which did not pass filtering criteria.
For additional information regarding all file formats used in ORFanage please consult the File Formats section.
Description of some important options¶
While default parameters of ORFanage are designed to be applicable to a wide array of tasks,
multiple optional parameters have been implemented to allow fine-tuning of the analysis.
Users can run orfanage --help or orfan -h to see all available parameters.
This section provides extended descriptions of some of the more important parameters.
--reference¶
Value specifies file with the genome sequence in FASTA format. Optional yet extremely useful parameter to include whenever possible.
Note
Using ORFanage without reference genome
Without a reference genome provided ORFanage is incapable of searching for novel start and stop codons, which limits utility to:
finding transcripts with identical ORFs to the reference
anotating all possible complete and partial ORFs. Results may include premature stop-codons.
Many other optional parameters can still be used without reference genome, yet their utility will be limited.
For instance, ILPD metric will still be computed based on the interval intersection of the reference ORF and query transcript.
Thus filtering via --ilpd <val> can still be used to retain only results passing a minimum ilpd threshold.
However, such filtering will not consider any novel sequence required to complete the reported ORF.
Reference genome should be provided whenver searching for all novel complete ORFs.
If the goal of the analysis is to
--stats¶
This parameter enables additional outputs with extended information regarding possible ORFs. The value sets the output filename to which extended information will be written. Please refer to File Formats. for detailed explanation regarding the contents of the stats file.
--mode¶
The value sets the scoring function used to compare candidate ORFs and select the best one. Possible values are:
START_MATCH - Selects the ORF candidate which matches the reference START codon.
LONGEST_MATCH - Selects the ORF candidate, which maximizes the number of positions shared between reference and query in the same frame. If alignment mode is enabled via
--pithis mode will be superceeded by the number of aligned positions instead.BEST - Default. Selects the ORF candidate, which maximizes the ILPI between reference and query. If alignment mode is enabled via
--pithis mode will be superceeded by highest % Identity instead.LONGEST - Selects the longest ORF candidate.
ALL - reports all available ORF candidates.
The default order in which ORFanage will evaluate candidate ORFs is BEST,START_MATCH,LONGEST_MATCH,LONGEST,ALL
The method iterates over the metrics in the specified order, elimitating non-passing candidate ORFs until a single candidate ORF remains.
--use_id¶
Using this flag enables grouping and processing of transcripts by gene ID instead of relying only on overlaps between transcripts.
In most cases, we expect query datasets to have come from transcriptome assembly pipelines without properly assigned gene IDs that would match the template annotation. To detect which template ORFs to compare against the query transcripts, ORFanage will use simple coordinate overlaps. However, in some cases, such as polycistronic genes, miscRNA, etc simply overlapping reference with query will results in undesired comparisons. Suppose…
If compatible gene ID assignments are available for query and refernece datasets, users may wish to turn this flag on to limit comparisons to within members of the same gene only.
--threads¶
The value specifies the number of threads that ORFanage will be allowed to use concurrently. Internally, the method creates groups of transcripts based on coordinate overlap or gene ID and processes each group independently in a separate thread. This means ORFanage utilize efficiently roughly as many threads as there are genes.
--non_aug¶
This flag enables reporting of transcripts with non-canonical start codons. While translation of mRNA usually starts with an AUG codon, non-canonical translation initiation sites are known and documented. By enabling this flag, ORFanage may report ORFs with non-AUG start codons if and only if:
One of the template ORFs has a non-canonical start codon
The start of the best ORF reported by ORFanage matches coordinates of one of the reference non-canoniacl start codons perfectly.
--keep_cds¶
This flag prevents ORFanage from alterning any of the ORFs already present in the query dataset. This option is useful when a fraction of the query dataset has already been processed by other methods and is to stay intact.
--pi¶
The value sets the minimum % Identity threshold. internally, ORFanage computes a metric we term ILPI (In-frame Length Percent Identity)
Setting a value other than 0 for --pi will enable Smith-Waterman alignment via KSW2 library [3]. Rather than removing filtering via the default ILPI metric
the % Identity is only computed for the best ORF to be reported by the method. This reduces the computational burden of a full-fledged global alignment,
yet is sufficient to evaluate homology between any mismatching segments of the query and reference ORFs.
Note
ILPI
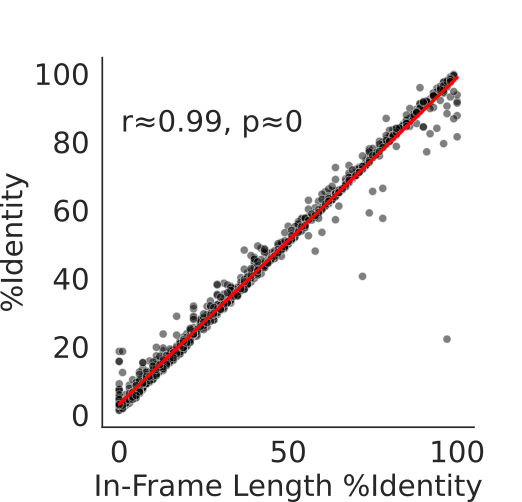
To compute ILPI, our method first computes the total number of positions in an ORF that are in the same frame as the reference, thus coding for the same codons, which determines the In-frame Length (IL). Then ILPI is computed as the fraction of IL of the total length of the reference coding sequence. As illustrated in the figure below, the correlation between ILPI and percent identity computed via the Smith-Waterman algorithm is very high.
Here illustrated is the the high correlation between ILPI and %Identity
--cleant¶
This flag enables cleaning of the reference annotation. Requires --reference value to be set. Some annotations include partial ORFs in attempt to increase robustness and comprehensiveness.
However having likely dysfunctioanal or incomplete data which may be undesirable, and --cleant helps streamline the process of removing any such cases.
When enabled, the software will assert the following properties for each reference transcript:
Each ORF starts with an AUG
Either the last 3 position of the ORF or the 3 positions on the transcripts that follow 3’ end of the ORF translate to a STOP codon
There are no in-frame STOP codons in the ORF
Any transcripts which fail to satisfy the above propertie are automatically discarded from comparisons.
--rescue¶
This flag instructs ORFanage to attempt fixing any reference ORFs that are invlaid or incomplete. The following operations will be performed on each ORF:
If an ORF does not start with an AUG - the ORF will be trimmed to the most upstream AUG within the ORF.
If no AUG is available with the ORF - the method will search upstream towards the 5’ end of the transcript
If an in-frame STOP codon is found - the 3’ end of the ORF is trimmed accordingly
If no STOP codon is found - the ORF is extended to the first available STOP codon upstream of the 3’ end of the transcript